An unpleasant hummingbird- A case of Multiple System Atrophy (MSA)

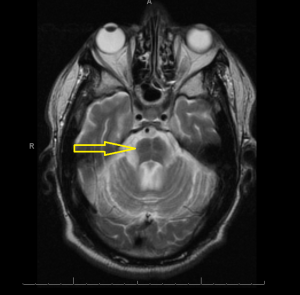
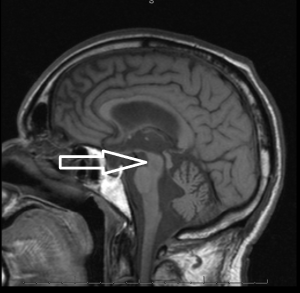
Middle age male patient admitted for evaluation of syncope. He has a history of coronary artery disease, severe pulmonary HTN, COPD, anemia of chronic disease, Osteoarthritis, left vocal cord paralysis, orthostatic hypotension, unsteady gaits and previous syncopal episode. CT head imaging remarkable for cerebellar atrophy. MRI done showed moderate cerebellar atrophy, midbrain atrophy with hummingbird sign and hot cross bun sign in the pons representing neurodegenerative disease multisystem atrophy and progressive supranuclear palsy.
The above constellation of symptoms and image findings are consistent with Multiple System Atrophy C-subtype.
Multiple system atrophy (MSA) is a sporadic [although mutations in the COQ2 have been implicated in some cases] neurodegenerative disease characterised by varying degrees of cerebellar ataxia, autonomic dysfunction, parkinsonism and corticospinal dysfunction. The estimated annual incidence of MSA in the population >50 years old is approximately 3 per 100,000. The estimated prevalence of MSA is between 2 to 5 cases per 100,000 population. Typically symptoms begin between 40 and 60 years of age.
The main clinical features of MSA are akinetic-rigid parkinsonism, autonomic failure including urogenital dysfunction, cerebellar ataxia, and pyramidal signs. Clinically, it typically presents in one of three patterns:
- Shy-Drager syndrome is used when autonomic symptoms predominate
- Striatonigral degeneration shows predominant parkinsonian features
- Olivopontocerebellar atrophy demonstrates primarily cerebellar dysfunction
The motor presentations of MSA are classified into two separate but overlapping clinical subtypes:
- MSA-C: predominance of cerebellar symptoms (olivopontocerebellar atrophy)
- MSA-P: predominance of parkinsonian signs and symptoms (striatonigral degeneration)
MSA-P is characterized by bradykinesia, rigidity, postural instability/fall, action tremor and myoclonus, hemiballism, chorea and dystonia.
The features of MSA-C involve predominant cerebellar dysfunction that manifests as gait ataxia, limb ataxia, ataxic dysarthria, and cerebellar disturbances of eye movements (including gaze-evoked nystagmus, impaired smooth pursuits with saccadic intrusion, and ocular dysmetria).
Other features common to both subtypes include dysautonomia with high rates of urinary dysfunction (83%) and symptomatic orthostatic hypotension (75%), Sleep and breathing abnormalities(laryngeal stridor) and cognative dysfunction. Raynaud phenomenon, and anxiety and depression have also been associated with MSA
Multiple systemic atrophy results from abnormalities of alpha-synuclein metabolism, resulting in intracellular deposition. It has been postulated that spreading of aberrant alpha-synuclein from neurons to glia through functionally connected networks leads to glial and myelin dysfunction and an inflammatory cascade that promotes secondary neurodegeneration.
Motor abnormalities seen in MSA-P are due primarily to neuronal loss and gliosis in the substantia nigra, putamen, caudate, and globus pallidus. One of the distinguishing features of MSA vs idiopathic Parkinson disease is the lack of dramatic and sustained response to levodopa in MSA.
The definite diagnosis of MSA is based upon postmortem pathology showing alpha-synuclein-positive glial cytoplasmic inclusions with neurodegenerative changes in striatonigral or olivopontocerebellar structures. The clinical diagnosis of possible MSA requires a sporadic, progressive, adult onset disease with either parkinsonism or cerebellar ataxia and at least one feature suggesting dysautonomia plus one additional supporting feature such as atrophy on image scan of putamen, middle cerebellar peduncle, pons, or cerebellum.
Magnetic Resonance Imaging (MRI) is the modality of choice – atrophy of putamen, middle cerebellar peduncle, or pons on MRI is supportive for possible MSA. MRI radiographic features include hot cross bun sign, atrophy of cerebellum and midbrain, putaminal rim sign.
There is no effective disease-modifying or neuroprotective treatment for MSA and the disease progresses relentlessly culminating in death usually within 10 years of diagnosis. Management is symptomatic.